New paper: Epigenetic Classification of Colorectal Cancers by scATAC-seq
Our paper on the epigenetic regulation for the molecular sunbtypes of colorectal cancers are now published on Cancer Discovery, where we systematically revealed the epigenetic basis of the well-known iCMS and CIMP classifications of colorectal cancers.
Significance: Our work revealed the epigenetic basis of the well-known iCMS and CIMP classifications of colorectal cancers. Moreover, interpatient minor similarities and major diversities of chromatin accessibility signatures of TF target genes can faithfully explain the corresponding interpatient minor similarities and major diversities of RNA expression signatures of colorectal cancers, respectively.

Link: https://aacrjournals.org/cancerdiscovery/article-abstract/doi/10.1158/2159-8290.CD-23-1445/735072/Single-cell-chromatin-accessibility-analysis
Colorectal cancer (CRC) forms a highly heterogeneous group of tumors classified into distinct genomic, epigenomic, and more recently, transcriptomic subtypes (ref 1). In 2015, based on the transcriptome profile of bulk tumor samples, the consensus molecular subtype (CMS) of CRC was established (ref 2). CRCs are classified into four CMS categories (CMS1-4) characterized by immune infiltration, WNT and MYC activation, metabolic deregulation, and mesenchymal infiltration, respectively. Recently, by analyzing single-cell RNA sequencing (scRNA-seq) data exclusively derived from CRC malignant epithelial cells, the intrinsic-consensus molecular subtypes (iCMSs) were identified (ref 3), where malignant cells were classified into iCMS2 and iCMS3. iCMS2 cells correspond to CMS2 CRCs, and iCMS3 cells could be further classified into microsatellite unstable (MSI) and microsatellite stable (MSS), which correspond to CMS1 and CMS3 CRCs, respectively. The (i)CMS classification systematically describes the heterogeneity among CRC tumors, covering a wide range of tumor biological features such as driver mutations, gene expression, tumor microenvironment, and clinical features. However, there are fewer studies related to the epigenetic heterogeneity of CRC, and the epigenetic features of different molecularly typed tumors as well as the epigenetic regulatory mechanisms of subtype-specific gene expression remain unclear, which is an urgent problem to be solved in this field.
On April 4, 2024, Cancer Discovery published our research article entitled “Single-cell chromatin accessibility analysis reveals the epigenetic basis and signature transcription factors for the molecular subtypes of colorectal cancers” from Prof. Fuchou Tang’s research group (BIOPIC/ICG, Peking University), in collaboration with Xin Zhou’s team (Peking University Third Hospital). Our study systematically reveals the epigenetic basis of molecular subtypes of CRC, which provides important insights for understanding the gene expression regulation mechanisms during the development of CRC, as well as potential screening and therapeutic targets.
Our study performed high-quality single-cell Assay for Transposase-Accessible Chromatin using sequencing (scATAC-seq) analysis of paracancerous normal tissues, benign adenomatous tissues, and tumor tissues from patients with CRC, with a total of 80 samples collected from 29 patients covering all bowel segments from the cecum to the rectum. By analyzing the chromatin accessibility landscapes of CRC epithelial cells, the study made the following five major findings:
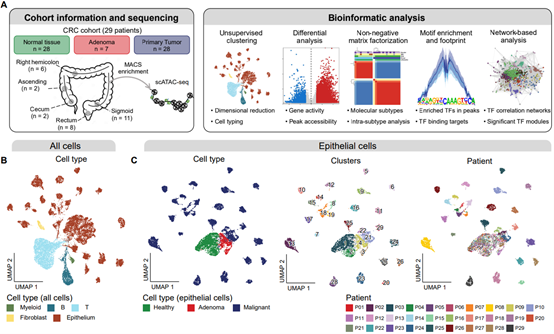
Figure 1. The chromatin accessibility landscape of CRC epithelial cells
1. Our study found that DNA methylation on tens of thousands of regulatory elements in the genome and corresponding chromatin accessibility were altered during the transition from healthy colorectal epithelium to benign adenomas and that most of these aberrant alterations in epigenetic status were stably inherited in subsequent malignant transformation of benign adenomas into adenocarcinomas.
Most CRCs develop from benign adenomas (ref 4). Our study found that chromatin accessibility was altered on tens of thousands of genomic cis-regulatory elements during the transformation of early normal intestinal epithelial cells into benign adenomas and that most of the newly acquired aberrant chromatin status in these adenomas was retained in CRCs. Notably, the chromatin peaks that were turned off in benign adenomas were significantly enriched in CpG islands, suggesting a possible synergistic effect of chromatin accessibility and DNA methylation. Through analysis of public DNA methylation microarray data, Our study confirms that abnormally closed chromatin peaks in adenomas are accompanied by an abnormal increase in DNA methylation, whereas abnormally open chromatin peaks are accompanied by abnormal DNA demethylation. This epigenetic co-regulatory process alters the status of several key CRC drivers and suppressors. These results suggest that during benign adenoma formation, chromatin accessibility and DNA methylation modification undergo tightly linked changes in the opposite direction, and that the two together regulate changes in the expression of key CRC driver and suppressor genes.
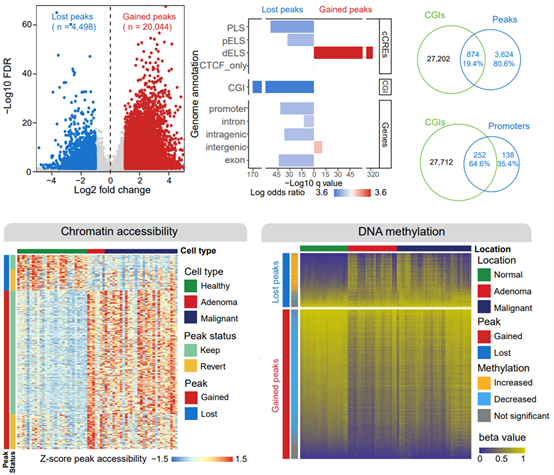
Figure 2. Chromatin dynamics during the generation of benign adenomas
2. Our study characterized the activity of key transcription factors that determine the most important molecular classification system (iCMS classification) of CRC, and found that only about 10% of the abnormally activated target genes within the same subtype were shared by different patients, while about 90% of the abnormally activated target genes were unique to different patients, which precisely revealed the epigenetic basis of shared and specific RNA expression characteristics of cancer cells in different patients. The epigenetic basis of common and individual RNA expression characteristics of cancer cells in different patients within the same subtype was precisely revealed.
To explore the epigenetic heterogeneity of CRC, Our study conducted an unsupervised molecular subtyping analysis of all CRC tumor cells and successfully identified two subtypes of cancer cells with different chromatin characteristics. The copy number variations, clinical traits, gene expression, and other characteristics of these two subtypes were highly consistent with the iCMS subtypes identified based on scRNA-seq data, confirming the consistency and robustness of the iCMS classification system at the epigenomic level. Our study also identified the genomic cis-acting elements and key transcription factors specifically activated in iCMS2 or iCMS3 subtypes and found that HNF4A and PPARA were specifically activated in tumors of the iCMS2 subtype, whereas FOXA3 and MAFK were specifically activated in tumors of the iCMS3 subtype. Further, by identifying target genes regulated by these transcription factors, Our study found that while these iCMS-specific transcription factors were co-abnormally activated in all tumors within the same subtype, the downstream target genes they regulated exhibited a high degree of patient specificity. Only about 10% of the aberrantly activated target genes were common to different patients, whereas about 90% of the aberrantly activated target genes were specific to different patients. These results suggest that aberrant activation of subtype-specific transcription factors is an important feature to distinguish different iCMS subtypes. Meanwhile, although tumors within the same molecular subtype share aberrant activation of the same transcription factors, the set of downstream genes regulated by them has strong inter-patient differences, and aberrant activation of transcription factors shapes both inter-subtypes and intra-subtype tumor heterogeneities.
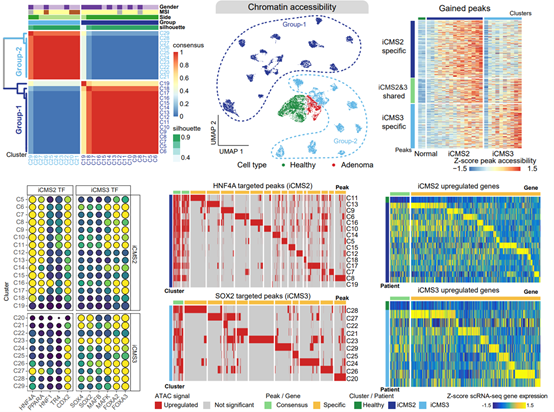
Figure 3. Cis-regulatory elements and transcription factors associated with iCMS classification
3. Through systematic analysis of intra-tumor subclones, Our study reveals the evolutionary mechanism of how cancer cells gradually acquire typical molecular subtype characteristics during tumor evolution.
In addition to inter-tumor heterogeneity, the study also systematically analyzed the epigenetic heterogeneities within tumors. Intra-tumor subclones with different chromosomal copy number variations were identified in seven CRC patients, with different subclones exhibiting different genome-wide chromatin accessibility landscapes. An in-depth analysis of P26 patients of the iCMS2 subtype revealed that subclones located downstream of the tumor evolutionary tree had higher iCMS2-specific transcription factor (e.g., HNF4A and PPARA) activities. Correspondingly, these subclones also exhibited higher iCMS2 module gene expression. These results reveal an evolutionary mechanism of how cancer cells gradually acquire typical molecular subtype characteristics during tumor evolution.
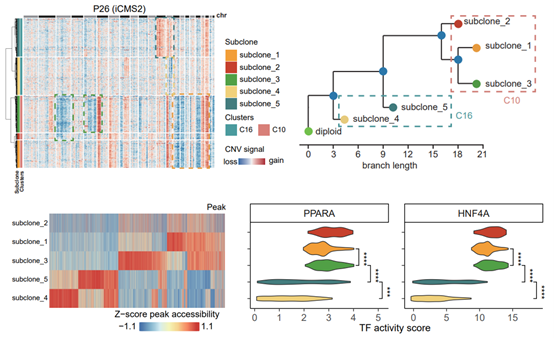
Figure 4. Epigenetic regulation of intra-tumor heterogeneity
4. Our study characterized the activity of key transcription factors that determine another important molecular classification (CIMP classification) system for CRC, and found that the chromatin status of cancer cells of the more malignant CIMP-Low and CIMP-Negative subtypes is much more abnormal than that of the less malignant CIMP-High subtype.
Some CRCs are characterized by aberrant hypermethylation of promoter CpG islands, a phenomenon known as the CpG-island methylator phenotype (CIMP, ref 5,6). In the chromatin accessibility atlas, Our study identified different CIMP subtypes and further identified the cis-regulatory elements and transcriptional factors that determine different CIMP subtypes. Compared to the more malignant CIMP-Low and CIMP-Negative isoforms, the CIMP-High subtype showed less abnormal chromatin status. Transcription factors LEF1 and TCF3 showed higher activity in the CIMP-High subtype, whereas the motif of the KLF family transcription factors was significantly enriched in the chromatin regions turned off in CIMP-High tumors, which may be related to the large-scale aberrant increase in methylation at promoter regions and silencing of the corresponding genes in CIMP-High CRCs.

Figure 5. Identification and epigenetic regulators of CIMP subtypes
5. The study identified transcription factor modules and their downstream target genes that are closely related to the multi-dimensional tumor heterogeneities of CRCs, and found that different core transcription factors contribute to CRC tumorigenesis through synergistic effects.
Finally, the study analyzed the transcription factor activities in the CRC atlas using weighted correlation network and identified 13 highly correlated transcription factor modules. Several of these modules were significantly correlated with tumor features such as iCMS, CIMP subtype, microsatellite stability, and primary location. Through further analysis of the network structure within each module, Our study identified key transcription factors that are closely associated with multi-dimensional tumor heterogeneities. In addition, by correlating transcription factor module activity with gene expression levels, Our study also identified potential downstream genes of key transcription factor modules. These results provide the epigenetic basis for how multiple transcription factors act synergistically to co-regulate gene expression, which in turn affects the tumorigenesis and molecular phenotype of CRCs.
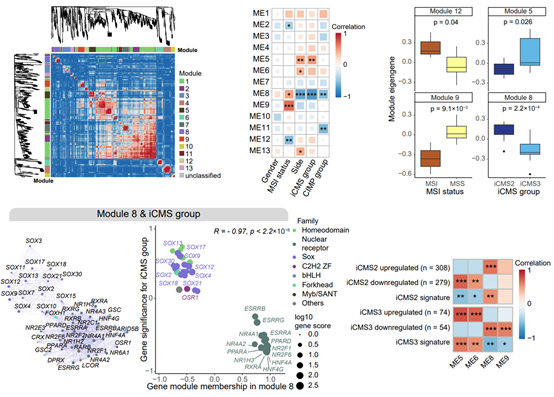
Figure 6. Transcription factor modules associated with multi-dimensional CRC heterogeneities
In summary, Our study systematically analyzed the chromatin accessibility landscapes of CRC epithelial cells. First, DNA methylation on tens of thousands of regulatory elements in the genome and the corresponding chromatin accessibility were altered during the transformation of normal colorectal epithelium to benign adenomas, and most of these aberrant alterations in epigenetic status were stably inherited during the subsequent malignant transformation of benign adenomas to adenocarcinomas. Secondly, the study characterized the activity of key transcription factors that determine the most important molecular classification systems (iCMS and CIMP classification) of CRCs, and found that only about 10% of the aberrantly activated target genes within the same iCMS subtype were shared by different patients, while about 90% of the aberrantly activated target genes were unique to different patients, which precisely revealed the common and individual characteristics of different patients within the same iCMS subtype. In addition, through the systematic analysis of intra-tumor subclones, Our study reveals the evolutionary mechanism of how cancer cells gradually acquire typical molecular subtype features during tumor evolution. Finally, Our study identified transcription factor modules and their downstream target gene sets that are closely related to the multi-dimensional tumor heterogeneity of CRCs, and found that different core transcription factors contribute to the tumorigenic process of CRCs through synergistic effects. These results deeply elucidate the epigenetic regulation mechanism of gene expression heterogeneities among CRC patients, and provide important clues for identifying potential screening and therapeutic targets for CRC.
Reference
Dienstmann, R. et al. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 17, 79–92 (2017).
Guinney, J. et al. The consensus molecular subtypes of colorectal cancer. Nat Med 21, 1350-1356 (2015).
Joanito, I. et al. Single-cell and bulk transcriptome sequencing identifies two epithelial tumor cell states and refines the consensus molecular classification of colorectal cancer. Nat. Genet. 54, 963-975 (2022).
Dekker, E., Tanis, P. J., Vleugels, J. L. A., Kasi, P. M. & Wallace, M. B. Colorectal cancer. Lancet 394, 1467–1480 (2019).
Weisenberger, D. J. et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 38, 787–793 (2006).
Toyota, M. et al. CpG island methylator phenotype in colorectal cancer. Proc. Natl Acad. Sci. USA 96, 8681–8686 (1999).
Zhenyu
at Peking University
2024.04.10